
How FDA Inspections Changed in 2026: What Investigators Are Actually Looking For Under QMSR

Introduction
The FDA's Quality Management System Regulation (QMSR) became mandatory on February 2, 2026, and the immediate question every medical device manufacturer should be asking is not "are we compliant?" but rather "what will the inspector actually look for when they walk through our door?"
The answer is: something fundamentally different from what you've experienced before. The FDA has not simply renamed the old Quality System Regulation and called it a day. They have retired the inspection methodology that served as the backbone of device inspections for over two decades and replaced it with a risk-based, ISO 13485-aligned framework that gives investigators significantly more latitude to follow the evidence wherever it leads.
In our Day 1 article on the QMSR transition, we covered the regulatory framework and what it means for your QMS at a strategic level. Today, we go deeper into the enforcement reality: how inspectors are trained under the new model, the "thread-pulling" techniques they use, and the five deficiencies that are already emerging as the most cited observations in early 2026 inspections.
At VigilaMed, we have guided manufacturers through 20+ successful audits with a 100% pass rate. That track record is built on understanding not just what the regulations say, but how inspectors apply them in practice. This article shares that insight with you.
The Old Playbook Is Gone: QSIT Retirement and What Replaces It
For over 20 years, FDA investigators followed the Quality System Inspection Technique (QSIT), a structured approach that divided every inspection into four fixed subsystems: management controls, design controls, corrective and preventive actions (CAPA), and production and process controls. Inspectors would sample from each subsystem using standardised tables, creating a relatively predictable experience for manufacturers who knew the format.
QSIT is now retired. In its place, the FDA has introduced Compliance Program 7382.850, an inspection framework purpose-built for the QMSR era. The difference is not cosmetic. Where QSIT was structured and predictable, the new model is risk-based, adaptive, and intelligence-driven.

Under the new framework, investigators assess compliance across six QMS areas:
| QMS Area | ISO 13485 Reference | Key Elements | | :--- | :--- | :--- | | 1. Management Oversight | ISO 13485 Clause 5 | Quality policy, management review, resource allocation, internal communication | | 2. Production & Service Provision | ISO 13485 Clause 7.5 | Process controls, validation, cleanroom management, servicing | | 3. Design & Development | ISO 13485 Clause 7.3 | Design inputs/outputs, verification, validation, transfer, change control | | 4. Change Control | ISO 13485 Clause 7.3.9 | Design changes, process changes, document change management | | 5. Outsourcing & Purchasing | ISO 13485 Clause 7.4 | Supplier qualification, quality agreements, incoming inspection | | 6. Measurement, Analysis & Improvement | ISO 13485 Clause 8 | CAPA, complaint handling, internal audit, statistical techniques |
Critically, investigators also assess compliance with four "other applicable FDA requirements" that sit outside ISO 13485: Medical Device Reporting (21 CFR 803), Corrections and Removals (21 CFR 806), Unique Device Identification (21 CFR 801 Subpart B), and Registration and Listing (21 CFR 807). These are uniquely American obligations that your ISO 13485 certification audit never touched.
The pivotal change is how investigators choose what to inspect. Under QSIT, the selection was formulaic. Under Compliance Program 7382.850, investigators use risk-based element selection, choosing which of the six QMS areas and sub-elements to examine based on the manufacturer's specific risk profile. This means two facilities making the same class of device could face entirely different inspection scopes depending on their adverse event history, complaint patterns, previous 483 observations, and CAPA track record.
The "Thread-Pulling" Approach: How Investigators Trace Connections Across Your QMS
Perhaps the most significant tactical shift for manufacturers to understand is the investigator's new emphasis on tracing connections across QMS subsystems—what we at VigilaMed call the "thread-pulling" approach.
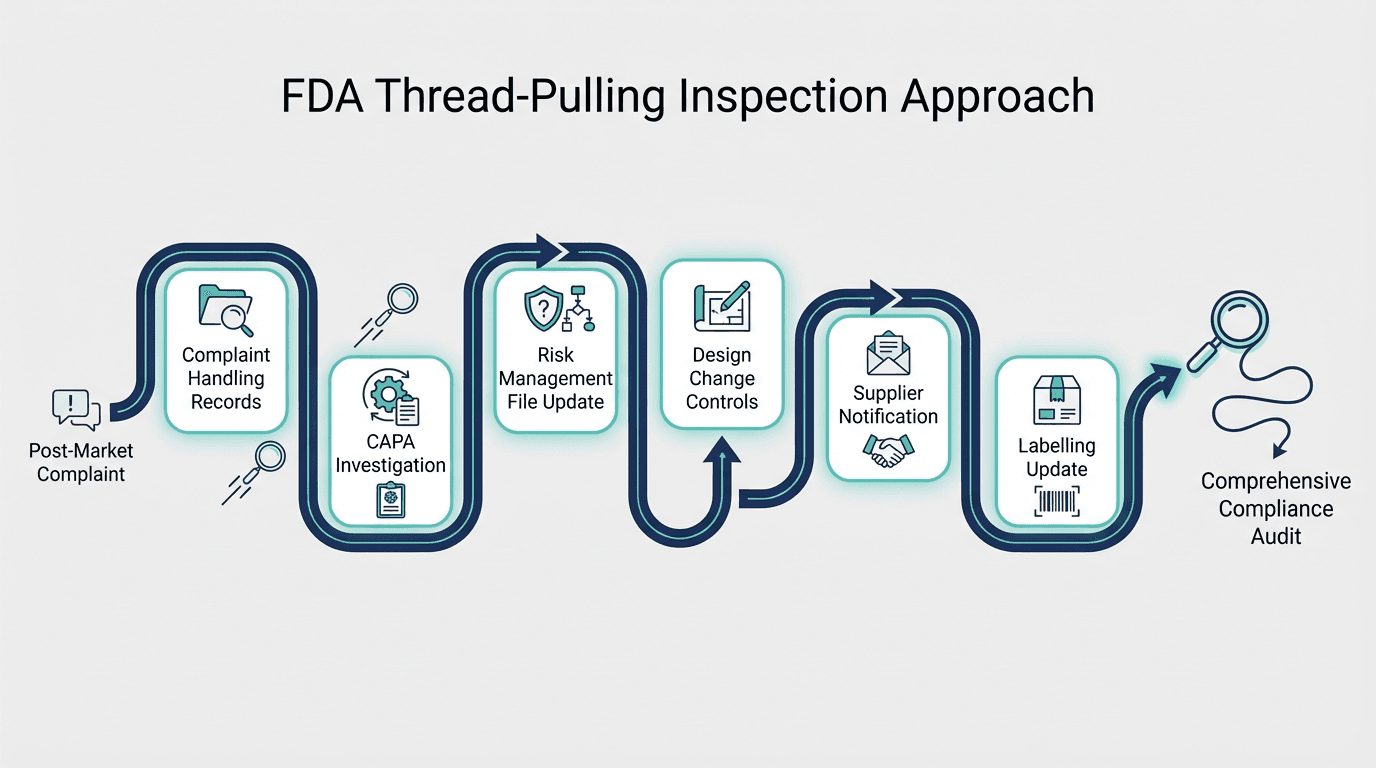
Under the old QSIT model, an investigator might examine your CAPA subsystem in relative isolation: were CAPAs documented? Were they completed? Under the QMSR model, the investigator is trained to pick a single thread—a specific complaint, adverse event, or CAPA record—and follow it through your entire quality system to test integration.
Here is a real-world example of how thread-pulling works in practice:
An investigator selects a post-market complaint about a device malfunction. They then trace the thread:
- Complaint handling records → Was the complaint properly investigated and trended?
- CAPA investigation → Was a root cause analysis conducted using a structured methodology?
- Risk management file → Was the risk file updated to reflect the new hazard data?
- Design change controls → Was a design change implemented to mitigate the risk?
- Supplier notifications → Were critical suppliers informed of specification changes?
- Labelling updates → Were instructions for use revised to address the identified hazard?
If any link in this chain is broken—if the CAPA exists but the risk file was never updated, or if the design change was validated but the supplier was never notified—the investigator has found a systemic integration failure that is far more serious than an isolated documentation gap.
This is a fundamentally different inspection philosophy. Under QSIT, a manufacturer could have gaps between subsystems that went unnoticed because each subsystem was examined independently. Under the QMSR model, the gaps between your systems are exactly where investigators are trained to look.
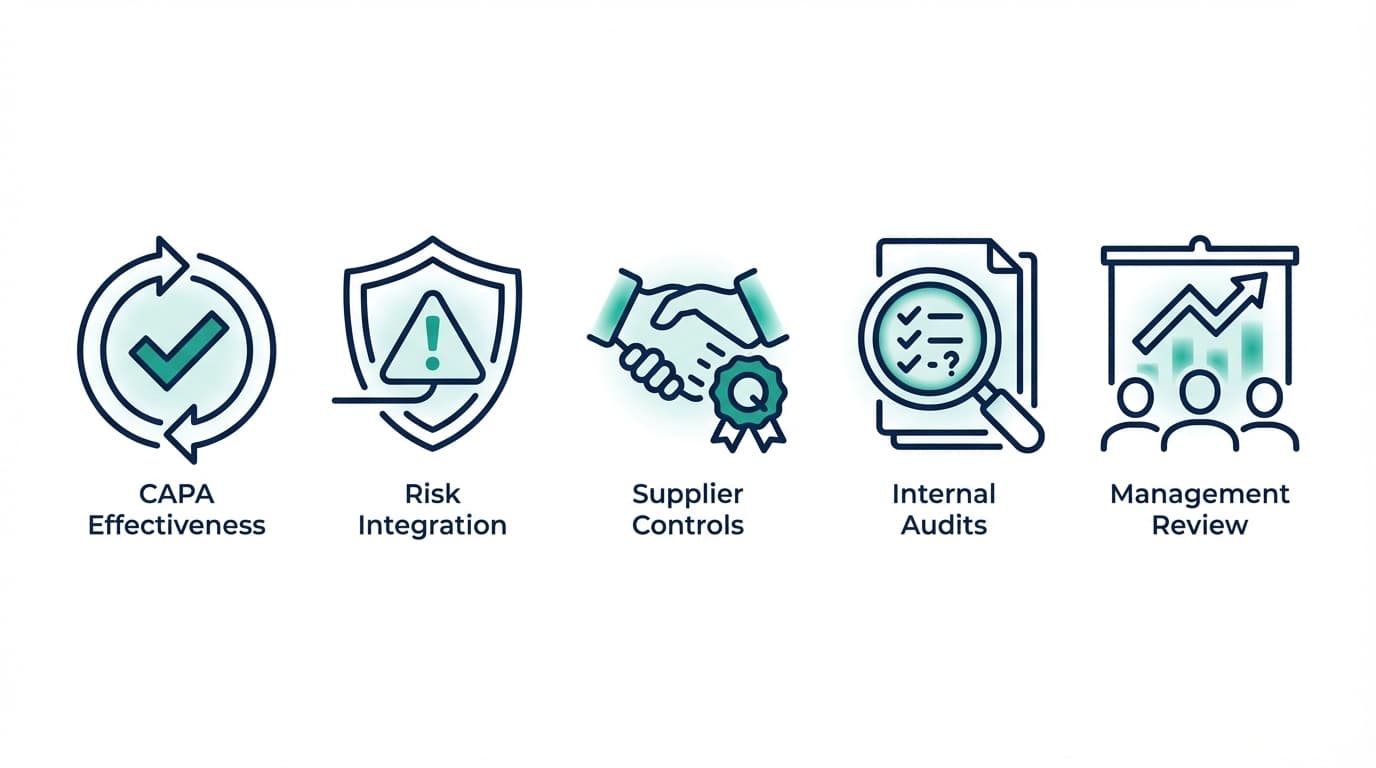
Top 5 Deficiencies FDA Is Citing Under QMSR: Early 2026 Inspection Data
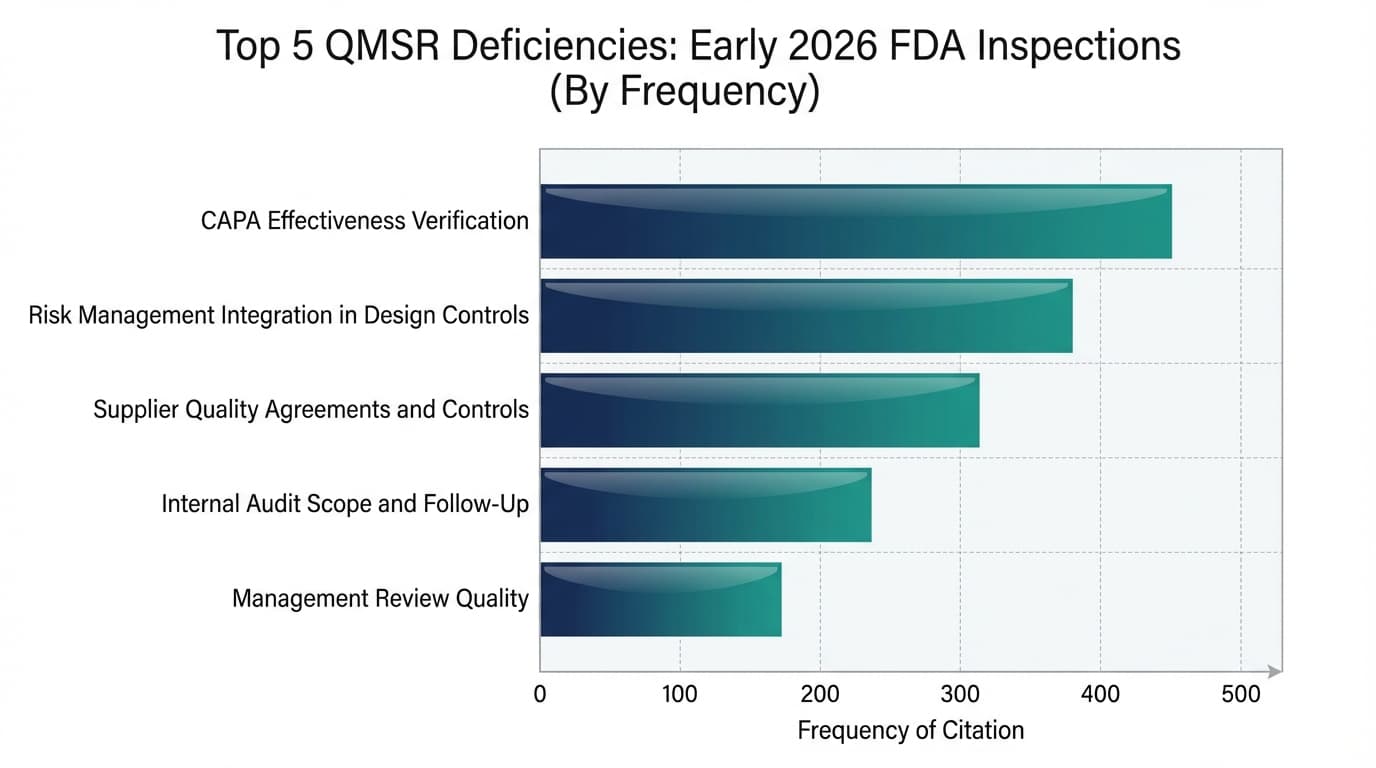
Based on analysis of early 2026 enforcement patterns, 483 observation trends, and intelligence from the regulatory community, five categories of deficiency are emerging as the most frequently cited observations under the new QMSR framework. These are not surprising to anyone who has been following FDA's enforcement trajectory—but the increased rigour and specificity of citations is new.

1. Inadequate CAPA Effectiveness Verification
CAPA has been the number one FDA citation for years, and under QMSR, the bar has been raised further. Investigators are no longer satisfied with CAPA records that show a corrective action was "completed." They want to see objective evidence that the corrective action actually worked.
What "good" looks like: Effectiveness verification should be based on measurable data—complaint trend analysis showing a decline in the specific failure mode after implementation, process metrics demonstrating improved yield, or audit findings confirming that the root cause has been eliminated. A sign-off sheet stating "CAPA verified effective" with no supporting data is a red flag.
2. Insufficient Risk Management Integration in Design Controls
The QMSR's explicit requirement for risk-based thinking throughout the QMS means investigators are scrutinising how risk analysis (ISO 14971) informs design decisions. Common citations include: design inputs that do not reference risk analysis outputs, verification and validation protocols that do not test against identified hazards, and design review records that do not demonstrate consideration of residual risk acceptability.
The question investigators are asking is: "Show me how your risk analysis influenced this design decision." If the answer requires shuffling through disconnected documents, you have a gap.
3. Weak Supplier Quality Agreements and Controls
Supplier management (ISO 13485 Clause 7.4) is receiving heightened scrutiny, particularly for critical components and contract manufacturers. Investigators are looking for documented quality agreements that clearly define responsibilities, acceptance criteria, change notification requirements, and audit rights. Informal email arrangements or purchase orders without quality clauses are being cited.
Additionally, investigators want to see that supplier audits are risk-based and proportionate—critical suppliers should be audited more frequently and more thoroughly than commodity suppliers. A one-size-fits-all supplier audit schedule is a vulnerability.
4. Incomplete Internal Audit Scope or Lack of Follow-Up
Under the previous QSR, internal audit reports were protected from routine FDA inspection. Under QMSR, they are now direct evidence of QMS effectiveness and subject to full FDA review. This is a seismic change.
Investigators expect to see internal audits that are risk-based (not merely clause-by-clause checklists), that identify meaningful findings (not perfunctory reports with zero non-conformances year after year), and that demonstrate robust corrective action with verified effectiveness for every finding. Internal audits that look like they were conducted to tick a box rather than to genuinely improve the QMS are being cited.
5. Management Review Records That Don't Demonstrate Data-Driven Decision-Making
Management review minutes are also now within FDA inspection scope. Investigators are looking for evidence that top management is actively using quality data to drive strategic QMS decisions.
Red flags include: generic minutes that could apply to any company in any year, absence of specific quality metrics (complaint rates, CAPA closure times, audit findings, process yields), no documented decisions or action items, and no evidence that previous action items were followed up. Effective management review should demonstrate that leadership is engaged, informed, and making data-driven decisions about quality system performance—not simply attending a meeting.
The New Scrutiny on Internal Audits and Management Reviews
It is worth emphasising just how significant this change in inspection scope is. Under the old QSR, 21 CFR 820.180(c) and 820.198(a)(1) protected management review records, internal audit reports, and supplier audit reports from routine FDA access. Manufacturers used this protection to encourage candid, no-holds-barred internal assessment.
That protection no longer exists. Under QMSR, which incorporates ISO 13485:2016 by reference, there is no equivalent shield. FDA investigators can—and will—request to see your internal audit reports, management review minutes, and supplier audit documentation.

The practical implications are significant:
| Record Type | What Investigators Expect | | :--- | :--- | | Internal Audit Reports | Must demonstrate risk-based audit planning, competent auditors, meaningful findings, effective corrective actions, and verified effectiveness. A history of audits with zero findings may actually raise suspicion rather than provide comfort. | | Management Review Minutes | Must reference specific quality data (complaint trends, CAPA metrics, audit results, customer feedback), document decisions taken, assign action items with owners and deadlines, and demonstrate follow-through on previous actions. | | Supplier Audit Records | Must show risk-based supplier selection, documented audit criteria, identified non-conformances, corrective action tracking, and evidence of re-evaluation. Supplier audits that are merely "tours of the facility" without documented assessment criteria are insufficient. |
AI-Driven Inspection Targeting: Why Your Next Inspection May Not Be Random
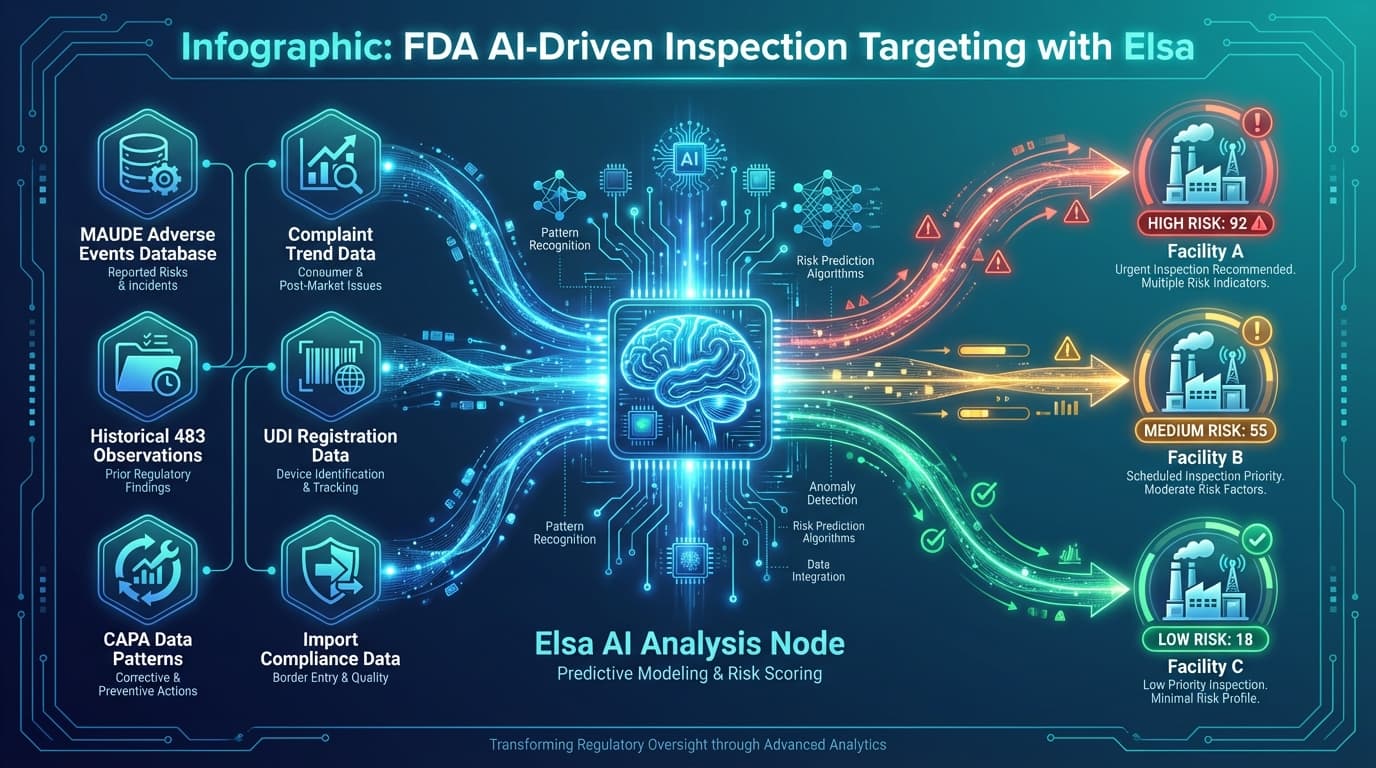
There is another dimension to the new enforcement reality that manufacturers need to understand: FDA inspections are becoming less random and more targeted. In June 2025, the FDA launched an internal advanced analytics system known as "Elsa," which analyses a vast array of data sources to identify facilities with elevated risk profiles.
The data sources feeding this system include:
- MAUDE adverse event reports — patterns of complaints and adverse events associated with your devices
- Historical 483 observations — unresolved or recurring issues from previous inspections
- CAPA data anomalies — patterns suggesting systemic quality issues
- Complaint data trends — complaint volumes, complaint-to-MDR ratios, trending patterns
- UDI and registration data — completeness and accuracy of device registration information
- Import data — for international manufacturers, patterns in import alerts and refusals
The strategic implication is clear: manufacturers with unresolved CAPA issues, recurring complaint patterns, or inconsistencies in their reported data are at significantly higher risk of inspection. The era of hoping you'll fly under the radar is over. Your data trail is your first impression, and the FDA is reading it before the investigator ever boards a flight.
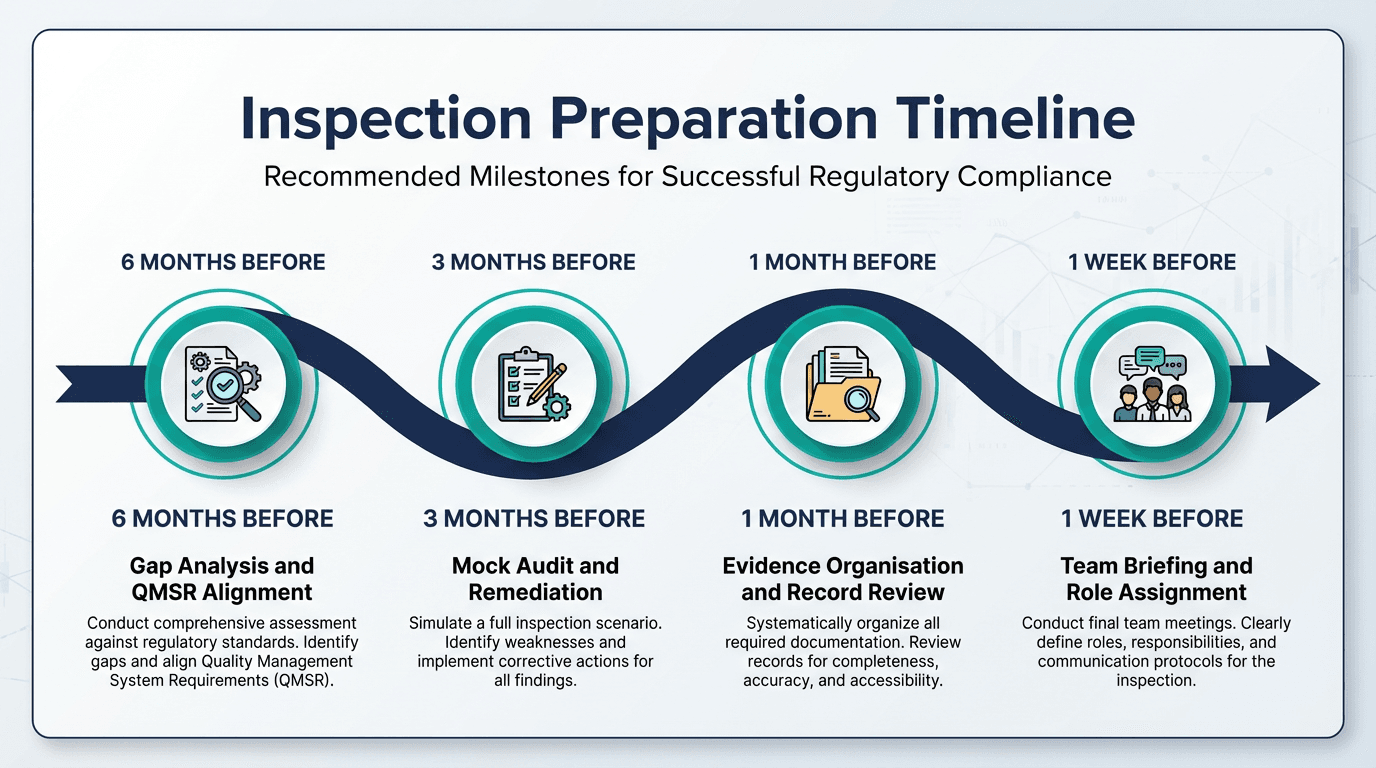
How to Prepare: The Mock Audit Advantage
Given the magnitude of these changes, manufacturers need a proactive preparation strategy. The most effective approach we recommend is a comprehensive mock FDA inspection conducted under the new QMSR framework.
A well-designed mock audit accomplishes four critical objectives:
-
Identifies systemic vulnerabilities before the real inspection reveals them. The thread-pulling approach often exposes integration gaps that internal teams overlook because they work within individual subsystems.
-
Tests evidence retrieval under realistic conditions. Can your team locate the management review minutes, the supplier audit report, and the CAPA effectiveness data within the timeframes an investigator expects? If not, the inspection will be painful even if the underlying documentation is sound.
-
Builds team confidence. Personnel who have experienced a simulated inspection understand how to interact with investigators, how to present evidence efficiently, and how to avoid the common mistake of volunteering information that was not requested.
-
Creates a remediation roadmap. A mock audit report provides a prioritised list of findings that your team can address systematically before the real inspection, ranked by risk and regulatory significance.

Frequently Asked Questions
Will FDA still use QSIT during inspections?
No. QSIT has been formally retired and replaced by Compliance Program 7382.850. All inspections conducted from February 3, 2026 onwards use the new risk-based, ISO 13485-aligned framework. Manufacturers should not expect the familiar four-subsystem QSIT format.
How long do QMSR inspections typically last?
Early indications suggest that QMSR inspections may run longer than typical QSIT inspections because the thread-pulling approach is more thorough. Where a routine QSIT inspection might last 3–5 days, QMSR inspections for medium-complexity manufacturers could extend to 5–8 days, depending on the risk profile and the number of threads the investigator chooses to follow.
Can I refuse to show internal audit reports?
No. Under the QMSR, the protections that previously shielded internal audit reports, management review minutes, and supplier audit records from routine FDA inspection have been removed. These records are now within the scope of FDA inspection, and refusal to provide them would be a serious compliance issue.
What if we have a CAPA backlog?
A CAPA backlog is one of the highest-risk signals to the FDA's AI-driven targeting system and to investigators during an inspection. If you have overdue CAPAs, prioritise them immediately based on risk to patient safety. Document your risk-based prioritisation rationale, and ensure that the CAPAs you are closing include robust effectiveness verification.
Conclusion and Next Steps
The transition from QSIT to the new risk-based inspection model represents more than a procedural change—it is a philosophical shift in how the FDA evaluates quality systems. Manufacturers who understand the thread-pulling methodology, prepare their records for expanded scrutiny, and proactively address the top deficiency areas will navigate this transition successfully.
Those who assume that passing previous QSIT inspections means they are ready for the QMSR era are taking a significant risk. The inspection model has changed, the scope has expanded, and the intelligence driving inspection targeting is more sophisticated than ever.
The time to prepare is now—not when you receive the FDA's notification of inspection.
Whether you need a mock FDA inspection to test your readiness under the new QMSR framework, a gap analysis to identify where your QMS falls short, or hands-on remediation support to close the gaps before the real inspection arrives, VigilaMed can help.
Schedule a Mock FDA Inspection — identify vulnerabilities before the real inspection arrives.
Book a Free Discovery Call to discuss your inspection readiness with a regulatory expert.
Already know what you need? Request a Proposal and we'll scope a project tailored to your timeline.
Need help with Regulatory News?
Our team of experts can help you navigate these regulatory requirements seamlessly. Book a discovery call today.
Book a Discovery Call